On the basis of their selectivity for PKCs and CAMKs, we chose to primarily focus on the 4-azaindole series of inhibitors, since they clearly displayed greater selectivity for PKD1 than the quinolinylmethylenethiazolinone derivative. 4-Azaindoles have previously been developed and characterized as inhibitors of p38a/b by Trejo et al.. Compounds 140 and 139 have been designed to reduce the oxidation at the 4-pyridyl nitrogen, a predominant site of metabolic oxidation. As a result of this modification, in vivo efficacy has been demonstrated for compounds 140 and 139 in an acute rat model of LPS-stimulated TNFa synthesis, providing favorable pharmacokinetic parameters for compound 140. Based on the desirable drug-like physical properties and promising PK/PD values, compound 140, and most likely 139, were deemed potent and selective orally available p38 inhibitors. These findings provide strong support for further development of the 140 and 139 series of analogs as drug/lead structures towards potent and selective PKD1 inhibitors, or dual PKD1/p38 inhibitors, with in vivo activity. Although a kinase profile reveals a few additional targets of 4-azaindoles, compound 140 in general displayed excellent selectivity as compared to the non-substituted 4,7-azaindoles, indicating there remains a distinct possibility to achieve greater selectivity Pazopanib through further medicinal chemistry modifications. On the other hand, although it is desirable to obtain sole selectivity for a single kinase, multitargeted protein kinase inhibitors tailored towards a small subset of kinases with distinct biological functions could be 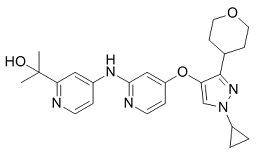 more attrY-27632 ROCK inhibitor active therapeutically; and, in fact, this strategy has proven to be an effective treatment in oncology. In this regard, PKD inhibitors with dual action on p38a might be equally attractive therapeutically, since both kinases have been implicated in inflammatory responses and cancer development. To further explore the mechanism of actions of these active PKD1 compounds, molecular modeling technologies were utilized to investigate putative binding modes. The threedimensional structure of PKD1 was built based on high-resolution crystal structures of homologues, and the catalytic domain, which consists of two lobes and an intervening linker, was well modeled. Subsequently, docking simulations were carried out, in which all ligands were docked into the putative ATP binding pocket of the kinase domain, and the resulting docking scores were relatively high. The interactions between the active lead compound 139 and the PKD1 kinase domain were further illustrated in detail. The modeling results are congruent with our experimental findings, demonstrating that these compounds are PKD1 inhibitors binding to the ATP site of kinase domain. The computational analyses provide additional insights into the possible molecular interactions and important binding residues of PKD1 and will prove useful in our future pharmacophore refinements. Influenza is one of the most common infectious diseases, affecting millions of people around the world every year. Occasionally, it causes a catastrophic pandemic such as the “Spanish flu” in 1918, which killed 30-50 million people worldwide. The most effective means of protection against influenza is vaccination; however, its effectiveness has been limited because etiological influenza A and B viruses constantly undergo antigenetic change. Moreover, the time needed to prepare a vaccine against a newly isolated influenza virus is more than half a year. This makes an emergency vaccine preparation against a pandemic influenza virus, such as the 2009 pandemic, difficult. However, as a vaccine alternative, several anti-influenza drugs have been developed.
more attrY-27632 ROCK inhibitor active therapeutically; and, in fact, this strategy has proven to be an effective treatment in oncology. In this regard, PKD inhibitors with dual action on p38a might be equally attractive therapeutically, since both kinases have been implicated in inflammatory responses and cancer development. To further explore the mechanism of actions of these active PKD1 compounds, molecular modeling technologies were utilized to investigate putative binding modes. The threedimensional structure of PKD1 was built based on high-resolution crystal structures of homologues, and the catalytic domain, which consists of two lobes and an intervening linker, was well modeled. Subsequently, docking simulations were carried out, in which all ligands were docked into the putative ATP binding pocket of the kinase domain, and the resulting docking scores were relatively high. The interactions between the active lead compound 139 and the PKD1 kinase domain were further illustrated in detail. The modeling results are congruent with our experimental findings, demonstrating that these compounds are PKD1 inhibitors binding to the ATP site of kinase domain. The computational analyses provide additional insights into the possible molecular interactions and important binding residues of PKD1 and will prove useful in our future pharmacophore refinements. Influenza is one of the most common infectious diseases, affecting millions of people around the world every year. Occasionally, it causes a catastrophic pandemic such as the “Spanish flu” in 1918, which killed 30-50 million people worldwide. The most effective means of protection against influenza is vaccination; however, its effectiveness has been limited because etiological influenza A and B viruses constantly undergo antigenetic change. Moreover, the time needed to prepare a vaccine against a newly isolated influenza virus is more than half a year. This makes an emergency vaccine preparation against a pandemic influenza virus, such as the 2009 pandemic, difficult. However, as a vaccine alternative, several anti-influenza drugs have been developed.